Желтуха при галактоземии и муковисцидозе носит характер
Содержание статьи
АнатомияАнглийский язык тестированиеБилеты ГИМСВедомственная охранаВоспитание и обучение тестыГеографияДеление чиселКоординаты картыЛицензия на оружиеМедицинские тестыНовый годОбщественные науки ПословицыПрофтобор в военкоматеПсиходиагностикаРостехнадзорСпортивные тестыТаблица МенделееваТаблица умноженияТест MMPIТест на беременностьТест на ДальтонизмТест на темпераментТест РавенаТест тревожностиТесты для бухгалтеровТесты для госслужащихТесты для полицииТесты для спасателейТесты для судебных приставовТесты для учителейТесты ДОПОГТесты жизнестойкостиТесты на iqТесты на госслужбу РКТесты на профессиюТесты на статус адвокатаТесты по английскому языкуТесты по географииТесты по историиТесты по менеджменту и маркетингуТесты по охране трудаТранспортная безопасностьУголовное правоФизкультура и спорт тестыЧастная охранаЭкзамен для мигрантовЭкзамен ПДД 2015Экзамен ПДД 2016Экзамен ПДД 2018 онлайнЭкзамен ПДД онлайн 2017Экономика и предпринимательствоЭлектробезопасностьЮриспруденция, государство и право тесты
Вопрос из теста: Тесты по педиатрии 1601-2000 вопрос Сказать спасибо 322 Еще вопросы: Желтуха при неосложненной форме гемолитической болезни новорожденных исчезает к концу ________ жизни Первым признаком билирубиновой интоксикации у новорожденных детей является Патологией желудочно-кишечного тракта, наиболее часто встречающейся у новорожденных, перенесших асфиксию в родах, является Классом иммуноглобулинов, содержащихся в грудном молоке, обеспечивающих местный иммунитет кишечника у новорожденных детей, является Причиной нарушения становления биоценоза кишечника у новорожденного ребенка чаще всего является
|
Источник
елтуха у ребенка при муковисцидозе
Желтуха у ребенка при муковисцидозе
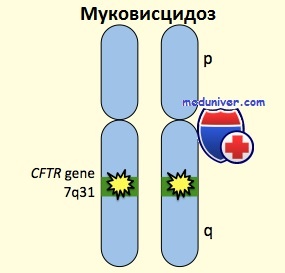
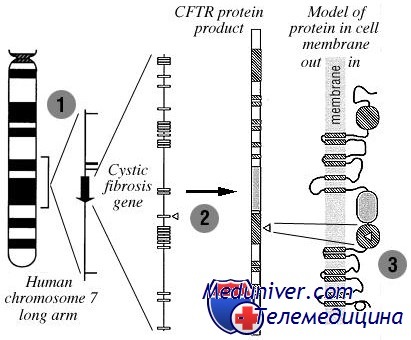
Муковисцидоз — это нарушение транспорта электролитов в эпителиальных клетках. Частично данную патологию можно диагностировать по повышенному уровню хлоридов в потовой жидкости. Поскольку аномальный транспорт электролитов через эпителий является основой патологических изменений при муковисцидозе, были проведены идентификация и клонирование гена в составе 7-й хромосомы, кодирующего CFTR.
Доказано, что CFTR является не только субстратом для активации фосфорилирования хлоридного канала, но и фактически самим хлоридным каналом, регулируемым циклическим аденозинмонофосфатом. К настоящему времени установлено, что муковисцидоз связан примерно с 1000 мутациями гена CFTR, наиболее частой из которых является F508 (трехосновная делеция, замещающая фенилаланин на 508-м месте).
В США мутация F508 присутствует более чем у 90% больных муковисцидозом. Механизм, посредством которого мутации CFTR вызывают заболевание, включает сокращение (или отсутствие) синтеза CFTR, дефект созревания белка, преждевременный распад, нарушенную регуляцию функции CFTR, дефект хлоридной проводимости или повышенное разрушение CFTR.
Хотя муковисцидоз более известен по нарушению секреции хлоридов в потовых железах и дыхательных путях, дефектные формы CFTR также ответственны за сгущение панкреатического сока и секретов гепатобилиарной системы. Поражение гепатобилиарной системы является наиболее частой внелегочной причиной летального исхода при муковисцидозе.
У новорожденных заболевание может иметь острые проявления в виде холестатической желтухи, особенно в тех случаях, когда у ребенка отмечался мекониальный илеус. В норме CFTR располагается вдоль апикальной зоны цитоплазматических мембран эпителиальных клеток желчных путей; в гепатоцитах его нет. Апикальная локализация CFTR в клетках желчных путей помогает понять, каким образом хлоридные каналы, регулируемые CFTR, участвуют в нормальной секреции желчи.
К классическим проявлениям муковисцидоза относят мальабсорбцию у младенцев в сочетании с повышенной частотой возникновения инфекций дыхательных путей в детском и подростковом возрасте. У многих новорожденных заболевание проявляется кишечной непроходимостью вследствие мекониального илеуса. Лишь у некоторых детей заболевание манифестирует клиникой неонатального холестаза, напоминающего атрезию желчных протоков.
У детей старшего возраста иногда диагноз «цирроз» или «портальная гипертензия» предшествует установлению диагноза «муковисцидоз». Многим пациентам с тяжелым поражением печени вследствие нарушения оттока желчи и последующего развития билиарного цирроза необходима трансплантация печени. Кроме оперативного лечения (т.е. трансплантации печени) единственно возможным консервативным методом терапии является лечение урсодеоксихолевой кислотой, хотя действие препарата при прогрессировании заболевания печени остается неясным.
С открытием факта, что CFTR локализуется в расположенных в подслизистом пространстве железах и поверхностном эпителии, эти локусы могут быть важны в плане разработки в будущем методов таргетной генной терапии. Таким образом, есть основания надеяться, что респираторные проявления муковисцидоза могут быть предотвращены переносом или экспрессией нормальной кДНК CFTR на эпителиальные клетки внутрипеченочных желчных протоков. Эти клетки желчных протоков могут быть исследованы путем применения аденовируса в качестве переносчика дефектного CFTR.
Данная потенциально превентивная терапия патологии печени, обусловленной муковисцидозом, с помощью генетически созданного CFTR в желчных протоках также дает возможность изучить и установить механизмы, ответственные за возникновение патологии гепатобилиарной системы у детей с муковисцидозом.
— Также рекомендуем «Желтуха у ребенка при тирозинемии»
Оглавление темы «Причины желтухи у детей»:
- Желтуха у ребенка при инфекции вируса простого герпеса
- Желтуха у ребенка при вирусном гепатите В
- Желтуха у ребенка при вирусном гепатите А, С, Д
- Желтуха у ребенка при ВИЧ
- Желтуха у ребенка при инфекции ЕСНО-вируса, парвовируса и аденовируса
- Желтуха у ребенка при дефиците альфа-1-антитрипсина (а1-АТ)
- Желтуха у ребенка при муковисцидозе
- Желтуха у ребенка при тирозинемии
- Желтуха у ребенка при неонатальной болезни накопления железа (НБНЖ)
- Желтуха у ребенка при врожденных нарушениях обмена желчных кислот
Источник
елтуха у новорожденных при нарушениях обмена веществ
Желтуха у новорожденных при нарушениях обмена веществ
Младенцы с тирозинемией (будет рассмотрена детально далее), галактоземией и фруктозурией (наследственной непереносимостью фруктозы) имеют схожие клинические проявления, особенно в первые дни и недели после рождения. Часто доминируют выраженная желтуха, гепатоспленомегалия, нарушения коагулограммы крови и ухудшение состояния ребенка в динамике.
У детей с галактоземией симптомы заболевания могут быть менее выражены, но по прошествии нескольких месяцев развиваются катаракта, цирроз печени и задержка психомоторного развития. Подобная ситуация может быть и у ребенка с тирозинемией, когда острая фаза заболевания отсутствует и лишь через несколько месяцев диагностируют цирроз, рахитические изменения костей и заболевание почек. Среди детей с этим заболеванием отмечена высокая частота опухолей печени.
Холестаз в сочетании с рвотой, отвращением к сладкой пище и фруктозурией, наиболее вероятно, является следствием непереносимости фруктозы. Каждое из указанных метаболических заболеваний может быть причиной дисфункции почек, которая проявляется аминоацидурией, глюкозурией и фосфатурией (синдром Фанкони). Более точная их диагностика основана на проведении специфических тестов на переносимость отдельных субстанций и определении активности фермента в эритроцитах (для галактоземии), печени или почках (в случае подозрения на непереносимость фруктозы), а также обнаружении сукцинилацетона в моче (при тирозинемии). Для начального скрининга подходит исследование мочи на нередуцирующие сахара, органические кислоты и аминокислоты.
При нарушении функции печени у новорожденного следует обязательно направить мочу и сыворотку крови в специальные лаборатории для проведения анализа на определение уровня первичных желчных кислот и промежуточных продуктов их метаболизма. На возможную болезнь накопления железа указывают маловодие или водянка плода в семейном анамнезе либо случаи смерти детей в первую неделю жизни. Исследование уровня ферритина в сыворотке, содержания железа в слюнных железах и печени (если проведение ее биопсии безопасно) помогает неонатологу быстро диагностировать это редкое нарушение метаболизма.
Персистирующая желтуха нетипична для младенцев с муковисцидозом, но она может наблюдаться, если это заболевание сопровождается мекониальным илеусом, гиперчувствительностью к лекарствам или частичной обструкцией общего желчного протока в результате сгущения желчи, а также если ребенок находится на парентеральном питании.

Дефицит а1-AT является наиболее частым нарушением метаболизма при патологии печени у младенцев. Обычно заболевание манифестирует развитием холестаза в сочетании с желтухой и гепатомегалией.
Диагноз подтверждается низким уровнем а1-АТ в сыворотке и определением ингибиторов протеазы (Pi) белка. Пока не существует специфического лечения, но считается перспективным использование генной терапии в будущем. Важно выявлять детей группы риска и проводить соответствующее генетическое консультирование в семьях. Согласно принятой тактике трансплантацию печени проводят детям более старшего возраста в терминальной стадии болезни печени, вызванной дефицитом а1-АТ. Прогноз при этом заболевании очень разнообразен.
Несмотря на наличие умеренно выраженной гепатоцеллюлярной дисфункции, через несколько месяцев после рождения может наступить клиническое улучшение. У некоторых детей в более старшем возрасте исходом заболевания могут стать билиарный цирроз и портальная гипертензия.
Наследственные болезни накопления, такие как болезнь Нимана-Пика и болезнь Гоше, обычно приводят к гепатоспленомегалии у младенцев; холестаз для этих болезней не типичен. Болезни накопления встречаются редко, поэтому не следует проводить их диагностику у детей с холестатической желтухой на начальном этапе диагностического поиска.
Полное парентеральное питание, как правило, приводит к поражению печени у новорожденных. ППП-ассоциированная гепатопатия обычно проявляется в виде холестаза, который проходит после отмены парентерального питания. У некоторых детей процесс восстановления печени может занять несколько месяцев, у других может развиться стойкое нарушение ее функции или печеночная недостаточность, приводящая к необходимости трансплантации печени.
Частыми факторами, влияющими на восстановление функции печени, являются сопутствующие инфекции (например, сепсис), хирургическая патология ЖКТ и недоношенность. Наличие этих факторов обусловливает недостаточную изученность патогенеза гепатопатии, ассоциированной с ППП. Основное лечение этого состояния заключается в отмене парентерального питания и разумном назначении энтерального кормления.
— Также рекомендуем «Синдромы Дубина-Джонсона, Ротора у новорожденного — клиника, диагностика»
Оглавление темы «Желтуха у новорожденных детей»:
- Атрезия желчных протоков у новорожденных детей
- Гипоплазия желчных протоков и киста общего желчного протока у новорожденных детей
- Неонатальный гепатит — клиника, диагностика
- Желтуха у новорожденных при нарушениях обмена веществ
- Синдромы Дубина-Джонсона, Ротора у новорожденного — клиника, диагностика
- Желтуха у новорожденных при бактериальных инфекциях — клиника, диагностика
- Желтуха у новорожденного ребенка при токсоплазмозе — клиника, диагностика
- Желтуха у новорожденного ребенка при сифилисе — клиника, диагностика
- Желтуха у новорожденного ребенка при врожденной краснухе
- Желтуха у новорожденного ребенка при цитомегаловирусной (CMV) инфекции
Источник
Галактоземия
Галактоземия — наследственная ферментопатия, характеризующаяся нарушением нормального процесса углеводного обмена, а именно — метаболизма галактозы. Признаками галактоземии являются непереносимость грудного молока и молочных смесей, рвота, анорексия, гипотрофия, желтуха, цирроз печени, спленомегалия, отеки, катаракта, задержка психомоторного развития. Скрининг на галактоземию проводится всем новорожденным; дополнительное обследование включает определение уровня галактозы в крови и моче, проведение нагрузочных проб с галактозой и глюкозой, генетическое тестирование, УЗИ брюшной полости, ЭЭГ и др. Основу терапии галактоземии составляет безлактозная диета, назначаемая с первых дней жизни ребенка.
Общие сведения
Галактоземия — наследственная патология обмена веществ, обусловленная недостаточностью активности ферментов, принимающих участие в метаболизме галактозы. Неспособность организма утилизировать галактозу приводит к тяжелым поражениям пищеварительной, зрительной и нервной системы детей в самом раннем возрасте. В педиатрии и генетике галактоземия относится к редким генетическим заболеваниям, встречающимся с частотой один случай на 10 000 — 50 000 новорожденных.
Впервые клиника галактоземии была описана в 1908 году у ребенка, страдавшего сильным истощением, гепато- и спленомегалией, галактозурией; при этом заболевание исчезло сразу после отмены молочного питания. Позднее, в 1956 г. ученый Герман Келкер определил, что в основе заболевания лежит нарушение метаболизма галактозы.
Галактоземия
Причины галактоземии
Галактоземия является врожденной патологией, наследуемой по аутосомно-рецессивному типу, т. е. заболевание проявляется только в том случае, если ребенок наследует две копии дефектного гена от каждого из родителей. Лица, гетерозиготные по мутантному гену, являются носителями заболевания, однако у них тоже могут развиваться отдельные признаки галактоземии в легкой степени.
Превращение галактозы в глюкозу (метаболический путь Лелуара) происходит при участии 3-х ферментов: галактоза-1-фосфатуридилтрансферазы (GALT), галактокиназы (GALK) и уридиндифосфат-галактозо-4-эпимеразы (GALE). В соответствии с дефицитом этих ферментов различают 1 (классический вариант), 2 и 3 тип галактоземии.
Выделение трех типов галактоземии не совпадает с порядком действия ферментов в процессе метаболического пути Лелуара. Галактоза поступает в организм с пищей, а также образуется в кишечнике в процессе гидролиза дисахарида лактозы. Путь метаболизма галактозы начинается с ее превращения под действием фермента GALK в галактозо-1-фосфат. Затем при участии фермента GALT галактозо-1-фосфат преобразуется в УДФ-галактозу (уридилдифосфогалактозу). После этого с помощью GALE метаболит превращается в УДФ — глюкозу (уридилдифосфоглюкозу).
При недостаточности одного из названных ферментов (GALK, GALT или GALE) концентрация галактозы в крови значительно повышается, в организме накапливаются промежуточные метаболиты галактозы, которые вызывают токсическое поражение различных органов: ЦНС, печени, почек, селезенки, кишечника, глаз и др. Нарушение метаболизма галактозы и составляет суть галактоземии. Наиболее часто в клинической практике встречается классический (1 тип) галактоземии, обусловленный дефектом фермента GALT и нарушением его активности. Ген, кодирующий синтез галактоза-1-фосфатуридилтрансферазы, находится в околоцентромерном участке 2-ой хромосомы.
Симптомы галактоземии
По тяжести клинического течения выделяют тяжелую, среднюю и легкую степени галактоземии.
Первые клинические признаки галактоземии тяжелой степени развиваются очень рано, в первые дни жизни ребенка. Вскоре после кормления новорожденного грудным молоком или молочной смесью возникает рвота и расстройство стула (водянистый понос), нарастает интоксикация. Ребенок становится вялым, отказывается от груди или бутылочки; у него быстро прогрессируют гипотрофия и кахексия. Ребенка могут беспокоить метеоризм, кишечные колики, обильное отхождение газов.
В процессе обследования ребенка с галактоземией неонатологом выявляется угасание рефлексов периода новорожденности. При галактоземии рано появляется стойкая желтуха различной степени выраженности и гепатомегалия, прогрессирует печеночная недостаточность. К 2-3 месяцу жизни возникают спленомегалия, цирроз печени, асцит.
Нарушения процессов свертывания крови приводит к появлению кровоизлияний на коже и слизистых оболочках. Дети рано начинают отставать в психомоторном развитии, однако степень интеллектуальных нарушений при галактоземии не достигает такой тяжести, как при фенилкетонурии. К 1-2 месяцам у детей с галактоземией выявляется двусторонняя катаракта. Поражение почек при галактоземии сопровождается глюкозурией, протеинурией, гипераминоацидурией. В терминальной фазе галактоземии ребенок погибает от глубокого истощения, тяжелой печеночной недостаточности и наслоения вторичных инфекций.
При галактоземии средней тяжести также отмечается рвота, желтуха, анемия, отставание в психомоторном развитии, гепатомегалия, катаракта, гипотрофия. Галактоземия легкой степени характеризуется отказом от груди, рвотой после приема молока, задержкой речевого развития, отставанием ребенка в массе и росте. Однако даже при легком течении галактоземии продукты обмена галактозы токсическим образом воздействуют на печень, приводя к ее хроническим заболеваниям.
Галактоземия может протекать в моносимптомной форме, при которой обнаруживается только поражение ЦНС, катаракта или диспепсические расстройства. Описан вариант бессимптомной (асимптоматической) галактоземии Дюарте, при которой недостаточность ферментов выявляется только при биохимическом исследовании крови.
Осложнения галактоземии включают цирроз печени, сепсис, кровоизлияния в стекловидное тело, первичную аменорею, синдром истощения яичников. При галактоземии у 50% детей дошкольного возраста выявляется моторная алалия, характеризующаяся трудностью организации и координации речевых движений, бедностью словарного запаса, обилием парафазий и персевераций при сохранном понимании обращенной речи.
Диагностика
Для снижения риска развития осложнений при галактоземии необходимо как можно более раннее выявление патологии. Возможна пренатальная диагностика галактоземии, включающая проведение биопсии хориона, амниоцентеза с последующим исследованием ворсин и амниотической жидкости. В России, согласно современным стандартам, осуществляется скрининг новорожденных на следующие наследственные заболевания: фенилкетонурию, врожденный гипотиреоз, галактоземию, адрено-генитальный синдром и муковисцидоз. Неонатальный скрининг проводится на 3-5 сутки у доношенных детей и 7-10 сутки — у недоношенных. С этой целью производится забор капиллярной крови, которая переносится на фильтровальную бумагу и виде высушенных пятен отправляется в генетическую лабораторию.
Если при неонатальном скрининге у ребенка выявляется подозрение на галактоземию, проводится повторное решающее тестирование. В случае повторного обнаружения высокого уровня галактозы в крови или низкого уровня исследуемого фермента, ребенку устанавливается диагноз галактоземии. Сведения о таком ребенке сообщаются участковому педиатру, а семья новорожденного приглашается на консультацию генетика в медико-генетическую консультацию. Врач-генетик проводит подробный анализ родословной, выполняет генетическое тестирование для выявления мутантного гена, объясняет специфику питания ребенка с галактоземией.
Иногда для диагностики галактоземии прибегают к определению уровня галактозы в моче, проведению нагрузочных проб с галактозой и глюкозой. Мониторинг биохимических показателей крови и общего анализа крови и мочи при галактоземии позволяет определить степень повреждения внутренних органов (почек, печени и др.).
Дети с галактоземией нуждаются в консультации детского невролога, детского офтальмолога, проведении электроэнцефалографии, УЗИ органов брюшной полости, биомикроскопии глаза. В некоторых случаях показана пункционная биопсия печени. Галактоземию следует дифференцировать от других гликогенозов, сахарного диабета I типа, врожденной атрезии желчных протоков, гепатита, гемолитической болезни новорожденных.
Лечение галактоземии
Основная роль в лечении галактоземии принадлежит диетотерапии. Особенность питания при галактоземии заключается в пожизненном исключении из рациона продуктов, содержащих лактозу и галактозу: любого молока (женского, коровьего, козьего, детских молочных смесей, низколактозных смесей и пр.), всех молочных продуктов, хлеба, выпечки, колбас, конфет, маргаринов и др. При галактоземии запрещается употребление растительных и животных продуктов, содержащих потенциальные источники галактозы — галактозиды (бобовые, соя) и нуклеопротеины (почки, печень, яйца и др.).
Дети, страдающие галактоземией, обеспечиваются специальными смесями на основе изолята соевого белка, гидролизата казеина, синтетических аминокислот, а также безлактозными казеинпредоминантными молочными смесями. С 4-х месячного возраста вводятся фруктовые и ягодные соки; с 4,5 месяцев — фруктовое пюре; с 5 месяцев — овощное пюре; с 5,5 месяцев — безмолочные каши из кукурузной, гречневой или рисовой муки в разведении специализированной смесью; с 6 месяцев — мясной прикорм на основе мяса кролика, цыпленка, индейки, говядины; с 8 месяцев — рыба. Альтернативным источником углеводов для пациентов с галактоземией служат продукты на основе фруктозы.
Для улучшения метаболических процессов назначаются поливитамины, кокарбоксилазу, АТФ, оротат калия. Лицам с галактоземией противопоказан прием спиртовых настоек и гомеопатических препаратов, поскольку последние содержат лактозу. Дети с речевыми нарушениями нуждаются в консультации логопеда и целенаправленной работе по коррекции ОНР.
Прогноз и профилактика галактоземии
Лечение галактоземии, начатое с первых дней жизни позволяет избежать развития цирроза, катаракты, олигофрении. Если лечение начато в более поздние сроки, когда уже произошло поражение печени и ЦНС, с помощью рациональной диетотерапии прогрессирование заболевания можно замедлить. При тяжелых формах галактоземии может быть летальный исход. Диспансерное наблюдение ребенка с галактоземией осуществляется педиатром, генетиком, диетологом, детским окулистом и детским неврологом. Детям с галактоземией присваивается инвалидность.
Учитывая наследственную обусловленность галактоземии, медико-генетическое консультирование рекомендуется пройти будущим родителям, в чьих семьях есть родственники или дети с данным заболеванием. Беременным с высоким риском рождения ребенка с галактоземией, следует ограничить употребление молочных продуктов.
Источник